









| arg | type | example | note |
|---|---|---|---|
| -json | boolean | 输出json格式结果 | |
| -save | boolean | 保存excel | |
| -academic | boolean | 学术使用,比如REVEL数据库注释 | |
| -hl | boolean | 使用耳聋变异库 | |
| -nb | boolean | 使用新筛变异库 | |
| -pp | boolean | 使用孕前变异库 | |
| -sf | boolean | 使用ACMG SF变异库 | |
| -acmg | boolean | 使用ACMG2015计算证据项PVS1, PS1,PS4, PM1,PM2,PM4,PM5 PP2,PP3, BA1, BS1,BS2, BP1,BP3,BP4,BP7 | |
| -autoPVS1 | boolean | 使用autoPVS1结果处理证据项PVS1 | |
| -allTier1 | boolean | 不进行tier1过滤 | |
| -allgene | boolean | tier1过滤不过滤基因 | |
| -cnvAnnot | boolean | 重新进行UpdateCnvAnnot | |
| -cnvFlter | boolean | 进行 cnv 结果过滤 | |
| -warn | boolean | 警告基因名无法识别问题,而非中断 | |
| -wgs | boolean | wgs模式 | |
| -couple | boolean | 夫妻模式 | |
| -trio | boolean | 标准trio模式 | |
| -trio2 | boolean | 非标准trio,但是保持先证者、父亲、母亲顺序 | |
| -redis | boolean | 使用redis服务注释本地频率 | |
| -redisAddr | string | 127.0.0.1:6380 | redis服务器地址 |
| -cfg | string | etc/config.toml | toml配置文件 |
| -geneId | string | db/gene.id.txt | 基因名-基因ID 对应数据库 |
| -list | string | sample1,sample2,sample3 | 样品编号,逗号分割,有顺序 |
| -gender | string | M,M,F | 样品性别,逗号分割,与 -list 顺序一致 |
| -qc | string | sample1.coverage.report,sample2.coverage.report | bamdst质控文件,逗号分割,与 -list 顺序一致 |
| -filterStat | string | L01.filter.stat,L02.filter.stat | 计算reads QC的文件,逗号分割 |
| -imqc | string | sample1.QC.txt,sample2.QC.txt | 一体机QC.txt格式QC输入,逗号分割,过滤 -list 内样品列表 |
| -mtqc | string | sample1.MT.QC.txt,sample2.MT.QC.txt | 线粒体QC.txt,逗号分割,过滤 -list 内样品列表 |
| -karyotype | string | sample1.karyotpye.txt,sample2.karyotype.txt | 核型信息,逗号分割 |
| -loh | string | loh1.xlsx,loh2.xlsx | loh结果excel,逗号分割,按 -list 样品编号顺序创建sheet |
| -lohSheet | string | LOH_annotation | sheet name |
| -kinship | string | kinship.txt | trio的亲缘关系 |
| -snv | string | snv1.txt,snv2.txt | snv注释结果,逗号分割 |
| -exon | string | sample1.exon.txt,sample2.exon.txt | exon CNV 输入文件,逗号分割,过滤 -list 内样品列表 |
| -large | string | sample1.large.txt,sample2.large.txt | large CNV注释结果,逗号分割 |
| -extra | string | extra1.txt,extra2.txt | 额外简单放入excel的额外sheets中 |
| -extraSheet | string | sheet1,sheet2 | -extra对应sheet name |
| -prefix | string | outputPrefix | 输出前缀,默认 -snv 第一个输入 |
| -log | string | prefix.log | log输出文件 |
| -tag | string | .tag | tier1结果文件名加入额外标签,[prefix].Tier1[tag].xlsx |
| -product | string | DX1516 | 产品编号 |
| -seqType | string | SEQ2000 | redis 查询关键词,区分频率库 |
| -specVarList | string | etc/spec.var.lite.txt | 特殊变异库 |
-wesim模式 输出.result.tsv时 额外 处理- 拆分
Zygosity - 额外 输出 两列:
Genotype of Family Member 1Genotype of Family Member 2
- 拆分
- 额外
Tier1统计 - 额外
Tier1判断规则 - 额外
遗传相符判断规则 - 与
-trio2相同- 额外
筛选标签标签规则 变异来源familyTag标签规则
- 额外
- 读取
annotation.Gene.transcript, 得到基因-转录本对应关系 - 读取
CNV数据文件列表到paths - 使用
addCnv2Sheet(sheet, title, paths, sampleMap, filterSize, filterGene, stats, key, gender, cnvFile)写入 sheet- 读取
paths到cnvDb,遍历item- 跳过其他样品编号
- 一体机模式,进行表头处理
anno.CnvPrimer进行引物设计item["OMIM_Gene"]作为 基因,获取geneIDs- 基于
geneIDs注释CHPO基因-疾病突变频谱 -cnvAnnot时- 转换
json格式写入item["CNV_annot"]
- 转换
item["OMIM_Phenotype_ID"]->item["OMIM"]-cnvFilter时exon cnv跳过item["OMIM"]为空large cnv跳过长度 < 1M
- 基于
title写入sheet row -wesim时- 基于
wesim.cnvColumn写入cnvFile
- 读取
- 解析
-extra和-extraSheet到extraArrayextraSheetArray - 校验
长度相等 - 遍历 ->
i- 识别
extraArray[i]后缀- 后缀
xlsx时,AppendSheetextraSheetArray[i]->extraSheetArray[i] - 其他时,作为
txt读取slice写入sheetextraSheetArray[i]
- 后缀
- 识别
- 写表头 “SampleID”
- 遍历
sampleList->sampleIDAddRow().AddCell().SetString(sampleID)sampleID写入 行
loadData读取到 哈希数值data- 文件名后缀识别 是否
gzip文件,分别读取
- 文件名后缀识别 是否
cycle1data循环1 ->itemannotate1(item)- inhouse_AF -> frequency
- 历史验证假阳次数 <- “重复数”
- score to prediction
- update Function
- update FuncRegion
- gene symbol -> geneID
- 基于
geneIDs注释CHPO基因-疾病突变频谱 - 注释 孕前数据库
- 注释 新生儿数据库
- 注释 耳聋数据库
- “Omim Gene” -> “Gene”
- “OMIM_Phenotype_ID” -> “OMIM”
-acmg时anno.UpdateSnv(item, *gender)- “引物设计”
item["flank"] += " " + item["HGVSc"]- “变异来源”
- 注释 Tier
- Tier1 时
anno.UpdateSnvTier1(item)anno.UpdateAutoRule(item)anno.UpdateManualRule(item)annotate1Tier1(item)
delDupVardata循环1 ->itemTier1时- “#Chr”+”Start”+”Stop”+”Ref”+”Call”+”Gene Symbol”+”Transcript” ->
key countVar[key] > 1时- 加入
duplicateVar[key]
- 加入
- “#Chr”+”Start”+”Stop”+”Ref”+”Call”+”Gene Symbol”+”Transcript” ->
- 遍历
duplicateVar->key,items- 最大
score = anno.FuncInfo[item["Function"]]->maxFunc - 遍历
items->itemscore < maxFunc时item["delete"] = "Y"deleteVar[key+"\t"+transcript] = truecountVar[key]--continue
- 最小
transcriptLevel[transcript]->minTrans
- 遍历
items->itemitem["delete"] != "Y"且transcriptLevel[transcript] != minTransitem["delete"] = "Y"deleteVar[key+"\t"+transcript] = truecountVar[key]--
- 最大
cycle2data循环1 ->itemTier1时anno.InheritCheck(item,inheritDb)- 统计 基因-转录本 维度 “Zygosity” “ModeInheritance” 分布计数
data循环2 ->item- “ClinVar Significance” 拼接 “CLNSIGCONF”
Tier1时- “MutationName” 记入
tier1Db - “#Chr”+”Start”+”Stop”+”Ref”+”Call”+”Gene Symbol”+”Transcript” ->
key - 非
deleteVar[key]annotate2(item)item["遗传相符"] = anno.InheritCoincide(item, inheritDb, *trio)item["遗传相符-经典trio"] = anno.InheritCoincide(item, inheritDb, true)item["遗传相符-非经典trio"] = anno.InheritCoincide(item, inheritDb, false)familyTag = "single"-trio || -trio2时familyTag = "trio"-couple时familyTag = "couple"
item["familyTag"] = anno.FamilyTag(item, inheritDb, "trio")item["筛选标签"] = anno.UpdateTags(item, specVarDb, *trio, *trio2)anno.Format(item)FloatFormat- 6 位精度
NewLineFormat<br/>->\n
-wesim时annotate2IM- “Gene Symbol” 属于
acmgSFGene时 item[“IsACMG59”] = “Y” - “DiseaseName/ModeInheritance” 拼接 “DiseaseNameCH”或”DiseaseNameEN”与”ModeInheritance”
-trio时,拆分 “Zygosity” 给 “Genotype of Family Member 1” “Genotype of Family Member 2”- 基于
wesim.resultColumn写入resultArray
- “Gene Symbol” 属于
- 写入 Tier1 “filter_variants”
- 非
-wgs时 写入 Tier2
- “MutationName” 记入
outputTier3时SteamWriterSetStringMap2Row(tier3SW, 1, tier3RowID, item, tier3Titles)- 流式写入
Tier3
- 流式写入
-wgs时wgsCycle- 初始化
wgsXlsx- MTTitle 写入 “MT” Sheet,filterVaristsTitle 写入 “intron” Sheet
data循环1 ->item- 重新计算
wgs模式Tier1 Tier1时anno.InheritCheck(item,inheritDb)- 统计 基因-转录本 维度 “Zygosity” “ModeInheritance” 分布计数
- 重新计算
data循环2 ->itemTier1时annotate4(item)item["遗传相符"] = anno.InheritCoincide(item, inheritDb, *trio)item["遗传相符-经典trio"] = anno.InheritCoincide(item, inheritDb, true)item["遗传相符-非经典trio"] = anno.InheritCoincide(item, inheritDb, false)-trio时item["familyTag"] = anno.FamilyTag(item, inheritDb, "trio")item["筛选标签"] = anno.UpdateTags(item, specVarDb, *trio, *trio2)- 写入 Tier2
- 额外(不在
tier1Db) 非 “no-change” 变异 - 写入
intronSheet
- 线粒体 写入 “MT” Sheet
- 初始化
-
trio
- 先证者有检出
- 自动化判断 不是 B/LB || 有证据项 PM2
- denovo
- 公共频率<=0.01
- 基因集
- 功能集
- Tier1
- ( WGS || SpliceAI Pred D ) && 非 “no-change”
- 本地频率<=0.01
- Tier1
- Tier2
- 本地频率<=0.01
- Tier2
- 功能集
- Tier2
- 基因集
- Tier2
- 公共频率<=0.01
- 非denovo
- 公共频率<=0.01
- 基因集
- 功能集
- Tier1
- ( WGS || SpliceAI Pred D ) && 非 “no-change”
- 本地频率<=0.01
- Tier1
- Tier3
- 本地频率<=0.01
- Tier3
- 功能集
- Tier3
- 基因集
- Tier3
- 公共频率<=0.01
- denovo
- 自动化判断 不是 B/LB || 有证据项 PM2
- 先证者有检出
-
single
- 自动化判断 不是 B/LB || 有证据项 PM2
- 公共频率<=0.01
- 基因集
- 功能集
- Tier1
- ( WGS || SpliceAI Pred D ) && 非 “no-change”
- 本地频率<=0.01
- Tier1
- Tier3
- 本地频率<=0.01
- Tier3
- 功能集
- Tier3
- 基因集
- Tier3
- 公共频率<=0.01
- 自动化判断 不是 B/LB || 有证据项 PM2
-
HGMD DM || ClinVar P/LP
- 非线粒体
- 公共频率 <= 0.05
- Tier1
- 非 Tier1
- Tier2
- 公共频率 <= 0.05
- 非线粒体
-
特殊位点库
- Tier1
- “splice-3”
- “splice-5”
- “init-loss”
- “start_lost”
- “alt-start”
- “frameshift”
- “nonsense”
- “stop-gain”
- “stop_gained”
- “span”
-
标签拼接,分号
;分割 -
Tag1AFThreshold = 0.05
-
tag1:
- 本地频率<=Tag1AFThreshold || 特殊变异库 || HGMD DM || ClinVar P/LP
- trio || trio2
- 遗传相符-经典trio == 相符
- 标签 T1
- 遗传相符-非经典trio == 相符
- 遗传模式: AR || XL || YL
- 标签 1
- 遗传模式: AD
- 纯合记录 均为 0
- 标签 1
- 纯合记录 均为 0
- 遗传模式: AR || XL || YL
- 遗传相符-经典trio == 相符
- single
- 遗传相符 == 相符
- 遗传模式: AR || XL || YL
- 标签 1
- 遗传模式: AD
- 纯合记录 均为 0
- 标签 1
- 纯合记录 均为 0
- trio || trio2
- 本地频率<=Tag1AFThreshold || 特殊变异库 || HGMD DM || ClinVar P/LP
-
tag2:
- 特殊变异库 || HGMD DM || ClinVar P/LP
- 标签 2
- 特殊变异库 || HGMD DM || ClinVar P/LP
-
tag3:
- 本地频率<=0.01
- 烈性突变
- 标签 3
- 烈性突变
- 本地频率<=0.01
-
tag4:
- 本地频率<=0.01
- PP3
- 标签 4
- PP3:
- 非 PVS1
- 保守性: {“GERP++_RS_pred”,”PhyloP Vertebrates Pred”,”PhyloP Placental Mammals Pred”}
- 无 不保守
- 至少2个 保守
- 有害性b1: {“Ens Condel Pred”,”SIFT Pred”,”MutationTaster Pred”,”Polyphen2 HVAR Pred”}
- 无 良性/多态
- 至少2个 有害
- PP3
- 有害性b2: {“dbscSNV_RF_pred”,”dbscSNV_ADA_pred”,”SpliceAI Pred”}
- 无 良性/多态
- 至少2个 有害
- PP3
- spliceAI 有害
- PP3
- 有害性b1: {“Ens Condel Pred”,”SIFT Pred”,”MutationTaster Pred”,”Polyphen2 HVAR Pred”}
- 至少2个 保守
- 无 不保守
- 保守性: {“GERP++_RS_pred”,”PhyloP Vertebrates Pred”,”PhyloP Placental Mammals Pred”}
- 非 PVS1
- 非重复区域 && 特定功能
- 标签 4
- 特定功能:
- stop-loss
- cds-ins
- cds-del
- cds-indel
- SpliceAI Pred == D && 功能非 no-change
- 标签 4
- PP3
- 本地频率<=0.01
appendLOHs(excel, lohs, lohSheetName, sampleList)- 遍历
lohs->i,path - 对应
sampleID = sampleList[i] AppendSheetlohSheetName->sampleID+"-loh"
parseQC-karyotype->karyotypeMap-qcloadQC(*qc, *kinship, qualitys, *wgs)kinship->kinshipHashsep="\t"-wgs时sep=": "
- 遍历
-qc->i,path- 遍历
paht->line^## Files : (\S+)-> “bamPath”sep分割,TrimSpace,填充quality[i]
-wgs时 修正 “bamPath”kinshipHash[quality[i]["样本编号"]]更新quality[i]
- 遍历
- 遍历
qualitys->qualityqualityKeyMap更新列karyotypeMap[quality["样本编号"]]更新 “核型预测”- “原始数据产出(Gb)” 更新 “原始数据产出(Mb)”
-wesim时- 基于
wesim.qcColumn写入qcFile
- 基于
-filterStat->loadFilterStat(*filterStat, qualitys[0])- 计算 “Q20 碱基的比例” “Q30 碱基的比例” “测序数据的 GC 含量” “低质量 reads 比例”
-imqc- 遍历
-imqc->path读入imqc - 遍历
qualitys->sampleID,qualityimqc[sampleID]更新qualityquality["Q20 碱基的比例"] = qcMap["Q20_clean"] + "%"quality["Q30 碱基的比例"] = qcMap["Q30_clean"] + "%"quality["测序数据的 GC 含量"] = qcMap["GC_clean"] + "%"quality["低质量 reads 比例"] = qcMap["lowQual"] + "%"
- 遍历
mtqc- 遍历
-mtqc->i,pathpath读入qcMapqcMap更新qualitys[i]
- 遍历
updateQC- “罕见变异占比(Tier1/总)”
- “罕见烈性变异占比 in tier1”
- “罕见纯合变异占比 in tier1”
- “纯合变异占比 in all”
- 循环 -> chr
- “chr”+chr+”纯合变异占比”
- “SNVs_all”
- “SNVs_tier1”
- “Small insertion(包含 dup)_all”
- “Small insertion(包含 dup)_tier1”
- “Small deletion_all”
- “Small deletion_tier1”
- “exon CNV_all”
- “exon CNV_tier1”
- “large CNV_all”
- “large CNV_tier1”
addQCSheetAddSheet("quality")- 遍历
qualityColumn->key横向赋值row=sheet.AddRowrow.AddCell().SetString(key)- 遍历
qualitys->qualityrow.AddCell().SetString(item[key])
| name | version | note |
|---|---|---|
| 全外疾病库 | 2023.Q1-2023.05.17 | |
| 基因突变谱 | V6.0.0.20230411 | |
| ACMGSF | V2.0.2023.5 | |
| PrePregnancy | PP155-V5.1.2_20230427 | |
| NBSP | V2.3.1.20230505 | |
| VIPHL | 20230509.30952 |
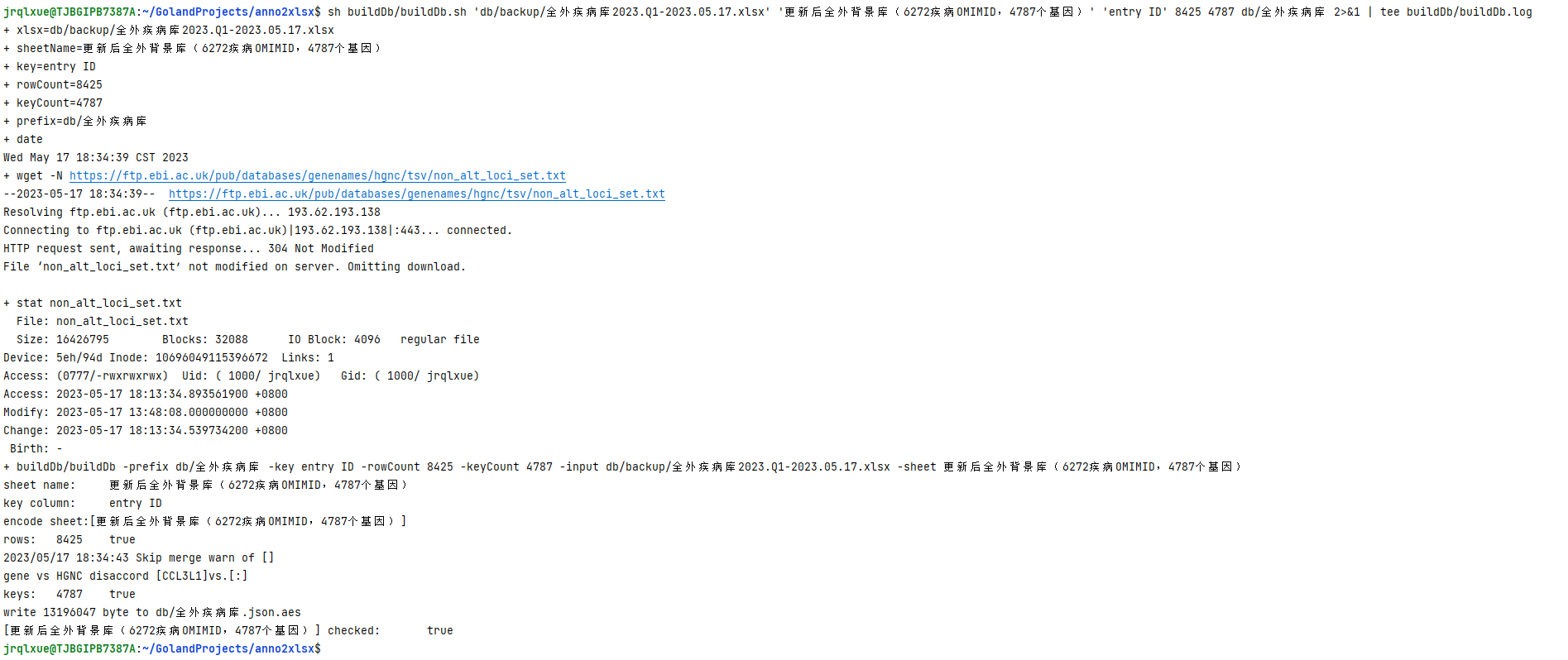
#!/bin/bash
wget -N https://ftp.ebi.ac.uk/pub/databases/genenames/hgnc/tsv/non_alt_loci_set.txt
stat non_alt_loci_set.txt
buildDb/buildDb \
-prefix db/全外疾病库 \
-key 'entry ID' \
-input db/backup/全外疾病库2023.Q1-2023.05.17.xlsx \
-sheet '更新后全外背景库(6272疾病OMIMID,4787个基因)' \
-rowCount 8425 -keyCount 4787
or
sh buildDb/buildDb.sh 'db/backup/全外疾病库2023.Q1-2023.05.17.xlsx' '更新后全外背景库(6272疾病OMIMID,4787个基因)' 'entry ID' 8425 4787 db/全外疾病库
sfCode=b7ea138a9842cbb832271bdcf4478310 # 替换成实际密钥
../NB2xlsx/buildDB/buildDB -code $sfCode -extract "#Chr,Start,Stop,Ref,Call,Gene Symbol,Transcript,cHGVS,证据项,致病等级,参考文献,关联疾病表型OMIM号,关联疾病英文名称,关联疾病中文名称,数据库时间" -input db/backup/ACMG\ 73基因23.05.23.xlsx -output db/ACMGSF.json.aes -keys etc/SF.key.txt
- 因本地无
WES注释流程,上传db/ACMGSF.json.aes和db/ACMGSF.json.aes.mut.tsv到集群 - 如本地有WES注释流程,所有操作均可在同一环境下完成
- 确认
致病性等级的 注释个数 一致
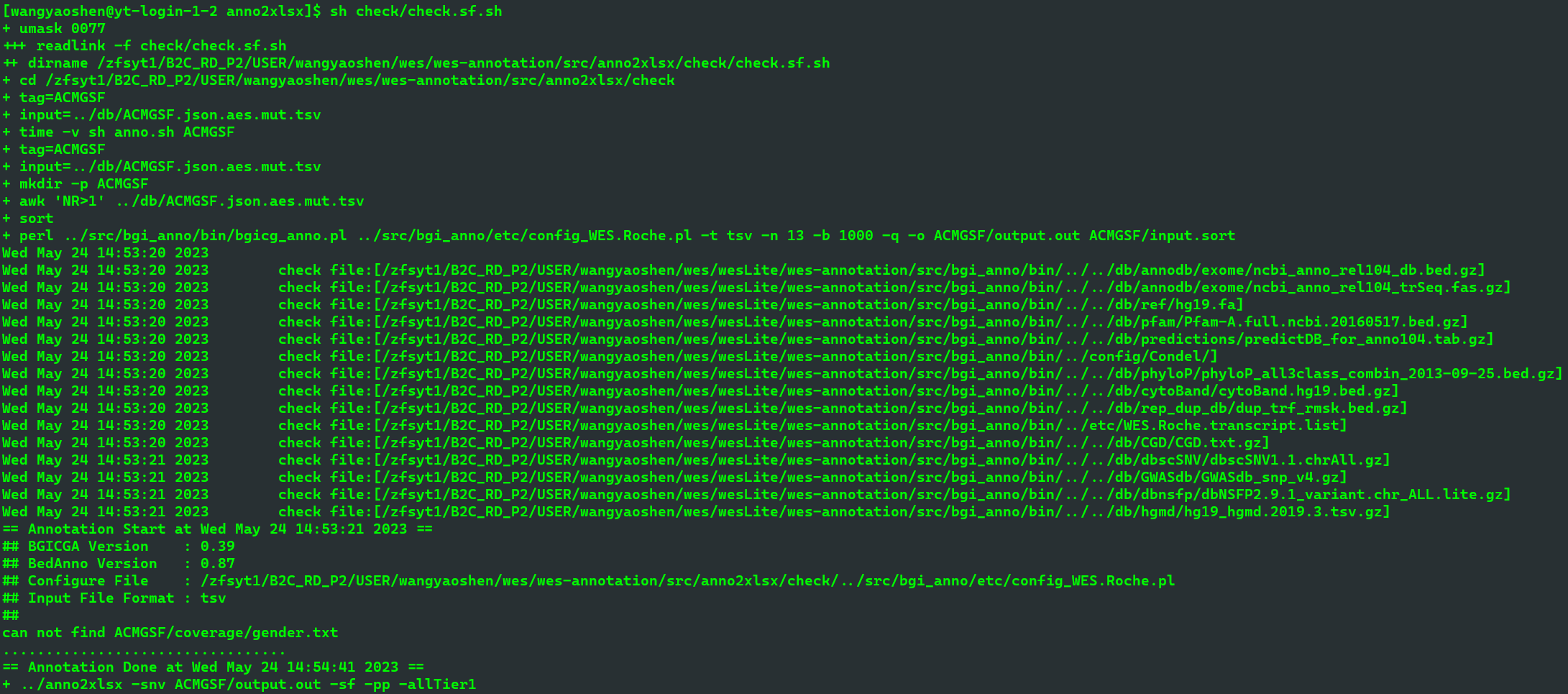
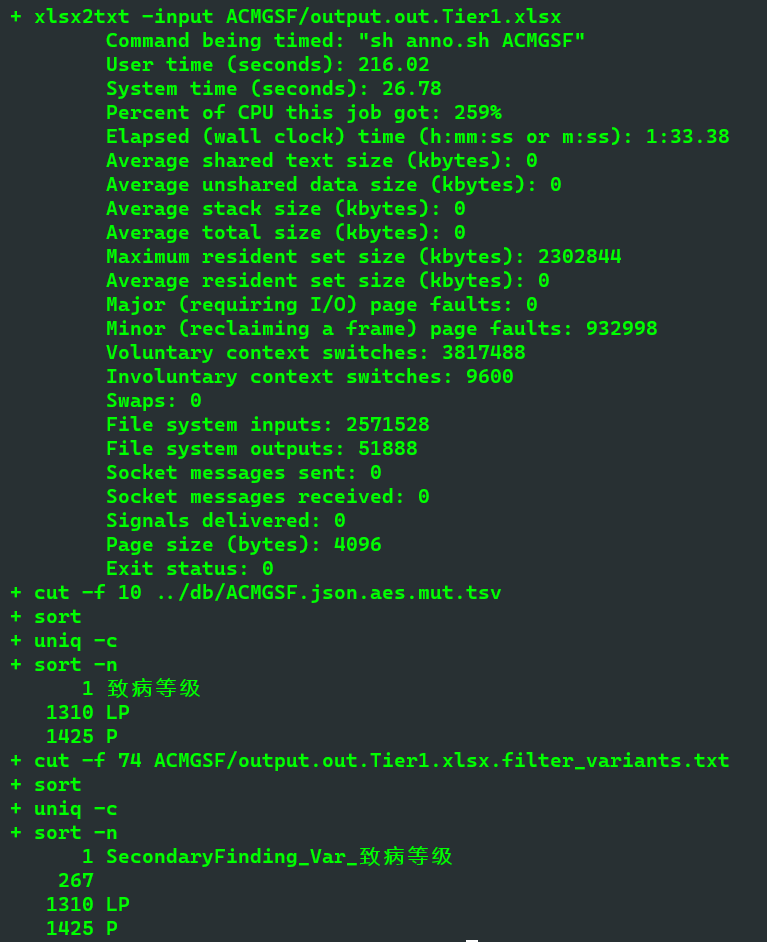
sh check/check.sf.sh

- 因原始
VIPHL库文件格式不同,需要转换格式步骤 - 因建库和注释校验均需
WES注释流程,整个下面示例步骤统一在集群环境处理,注意非加密文件的保密 - 如本地有
WES注释流程,建议在本地进行处理 - 核对
差异
## 生成 check/VIPHL/VIPHL.xlsx
chmod 700 check/viphl位点唯一结果统计_total_20230509.xlsx
sh check/build.hl.sh viphl位点唯一结果统计_total_20230509.xlsx
## 构建加密库
hlCode=6d276bc509883dbafe05be835ad243d7 # 替换成实际密钥
src/NB2xlsx/buildDB/buildDB -code $hlCode -extract "#Chr,Start,Stop,Ref,Call,Gene Symbol,Transcript,cHGVS,HLinterpretation,HLcriteria" -input check/VIPHL/VIPHL.xlsx -keys etc/HL.key.txt -output db/VIPHL.json.aes -sheet Result
## 注释校验
sh check/check.hl.sh


buildHPO -chpo chpo-2021.json -g2p genes_to_phenotype.txt -output db/gene2chpo.txt特殊位点库根据华大内部流程bgicg_anno.pl注释结果中的MutationName查找是否特殊位点,
所以以下情形可能发生库失效问题:
- 输入文件不包含以
MutationName命名的特定注释结果 - 输入文件
MutationName与流程bgicg_anno.pl结果不一致 - 流程
bgicg_anno.pl有更新,但是数据库未同步更新 - 位点用与现有配置不同的数据库注释导致的注释结果不一致
func zygosityFormat(zygosity string) string {
zygosity = strings.Replace(zygosity, "het-ref", "Het", -1)
zygosity = strings.Replace(zygosity, "het-alt", "Het", -1)
zygosity = strings.Replace(zygosity, "hom-alt", "Hom", -1)
zygosity = strings.Replace(zygosity, "hem-alt", "Hemi", -1)
zygosity = strings.Replace(zygosity, "hemi-alt", "Hemi", -1)
return zygosity
}func homRatio(item map[string]string, threshold float64) {
var aRatio = strings.Split(item["A.Ratio"], ";")
var zygositys = strings.Split(item["Zygosity"], ";")
if len(aRatio) <= len(zygositys) {
for i := range aRatio {
var zygosity = zygositys[i]
if zygosity == "Het" {
var ratio, err = strconv.ParseFloat(aRatio[i], 64)
if err != nil {
ratio = 0
}
if ratio >= threshold {
zygositys[i] = "Hom"
}
}
}
}
item["Zygosity"] = strings.Join(zygositys, ";")
}func hemiPAR(item map[string]string, gender string) {
var chromosome = item["#Chr"]
if isChrXY.MatchString(chromosome) && isMale.MatchString(gender) {
start, e := strconv.Atoi(item["Start"])
simpleUtil.CheckErr(e, "Start")
stop, e := strconv.Atoi(item["Stop"])
simpleUtil.CheckErr(e, "Stop")
if !inPAR(chromosome, start, stop) && withHom.MatchString(item["Zygosity"]) {
zygosity := strings.Split(item["Zygosity"], ";")
genders := strings.Split(gender, ",")
if len(genders) <= len(zygosity) {
for i := range genders {
if isMale.MatchString(genders[i]) && isHom.MatchString(zygosity[i]) {
zygosity[i] = strings.Replace(zygosity[i], "Hom", "Hemi", 1)
}
}
item["Zygosity"] = strings.Join(zygosity, ";")
} else {
log.Fatalf("conflict gender[%s]and Zygosity[%s]\n", gender, item["Zygosity"])
}
}
}
}因下游数据库结构新增字段开发工作量大,部分额外字段拼接进已有字段内
| 复用字段 | 拼接字段 | 连接字符串 | 是否可选拼接 |
|---|---|---|---|
ClinVar Significance |
CLNSIGCONF |
‘:’ | 是 |
flank |
HGVSc |
‘ ‘ | 是 |
- exon cnv输入文件不存在时仅log报错,不中断
WGS 使用anno2xlsx过滤后,进行spliceAI注释和过滤,然后重新进行Tier1判断、”遗传相符”和”筛选标签” 参考 README.md
- Tier1判断去冗余
Leave a Reply